What are cell and gene therapies and how are they different from drugs?
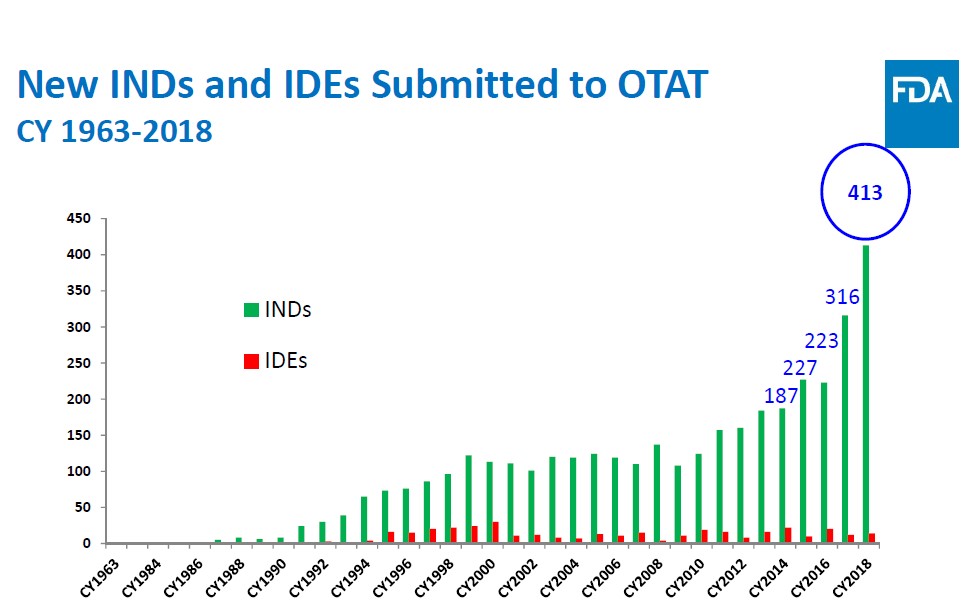
Cell and gene therapies are biologic drugs that come from living systems such as a microorganism or human cells (donors), whereas small-molecule pharmaceuticals largely come from combining specific chemical ingredients in an ordered process. The field of cell and gene therapy has gained significant attention over the last few years with Food and Drug Administration (FDA) approval of multiple cell therapy products (MACI, GINTUIT and PROVENGE) for treating particular diseases, as well as the first gene therapy approval (ZOLGENSMA) for children with spinal muscular atrophy. Indeed, the number of investigational new drug (IND) applications — program companies use to obtain permission to start a human clinical trial — sent to the FDA’s Office of Tissues and Advanced Therapies (OTAT) has nearly doubled in the last three years (Figure 1). Although these therapies have vast potential to treat or cure genetic disorders, autoimmune diseases and cancer, the scientific community and health authorities are only now starting to discover how novel cell and gene therapy products work, how to administer them safely and how to best attune the manufacturing process so that it meets demand within a reasonable time frame.
Given the continued need for a global open forum to discuss manufacturing, quality and regulatory considerations that arise from developing these products, the professional scientific society CASSS held the second annual Cell and Gene Therapy Products (CGTP) June 10–12, 2019. (Read more about CASSS and a synopsis of last year’s conference on the blog post Advancing the Manufacturing of Human Cell and Gene Therapies.) This year, Heidi Zhang joined Bruce Thompson and Andrew Weiskopf in co-chairing the three-day symposium at the Hyatt Regency hotel in Bethesda, Maryland. There were 245 attendees (172 were first-time attendees), representatives from 15 countries and 87 companies, 18 regulators and 30 speakers. The CASSS mobile app allowed attendees to access the schedule, presentation slides and speaker abstracts, and to network with fellow attendees.
What programs are available to expedite drug development?
To kick off the conference, Celia Witten, deputy director at the Center for Biologics Evaluation and Research (CBER) Office of the Center Director, provided the FDA perspective on development of cell and gene therapy products. First, she showed information on how the INDs submitted for CGTP increased by approximately 50% from 2017 to 2018. She then showed a figure on 34 regenerative medicine advanced therapy (RMAT) designations that were granted through May 1, 2019. Some rejections occurred due to products not meeting the RMAT designation, which is why it’s imperative to understand RMAT eligibility and all regulatory approaches for making CGTP rapidly available. Briefly, a drug is eligible for RMAT designation if:
a. The drug is a regenerative medicine therapy (e.g., cell therapy).
b. The drug is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition.
c. The drug, through evidence shown by preliminary clinical data, has potential to address unmet medical needs for a disease or condition.
Steven Oh, deputy director at the CBER (which is at the FDA’s OTAT), followed Witten’s talk and discussed expedited development programs — accelerated approval, priority review, fast track, breakthrough therapy (BT) and RMAT — in detail, as well as the regulatory chemistry, manufacturing and control (CMC) considerations behind them. For instance, he mentioned that clinical programs tend to move at a fast rate for BT and RMAT products because the early to late development stages are condensed. However, a rapid clinical development does not change the CMC requirements. Therefore, a high focus should be placed on all CMC and current good manufacturing practices (CGMPs) issues as early as possible if products receive BT or RMAT designation. If the CMC requirements could be met as quickly as clinical development, it would help ensure the availability of a high quality product that can be steadily made at the time of approval. Although the type and extent of manufacturing information expected at the time of submission is evaluated on a case-by-case basis, the FDA representatives pointed out that there are some areas of potential flexibility, such as validation strategies, manufacturing scale-up/scale-out strategies, and use of post-marketing commitments or requirements.
What are neoantigens and how are they made?
An interesting topic introduced during the conference’s second day was individualized neoantigen-specific cancer vaccines. Unlike traditional vaccines intended to prevent diseases such as polio or measles directly, cancer vaccines do not directly attack the disease but rather help the immune system build antibodies to recognize and fight off cancer. There are two types of cancer vaccines: preventive and treatment. Preventive vaccines work by killing viruses that may lead to cancer, while treatment vaccines are designed to stimulate the immune system to attack cancerous cells.
Antigens are substances on the surface of cells that are not normally part of the body. Normally, the immune system attacks the antigens and usually gets rid of them, leaving the immune system with a “memory” that helps it respond to those antigens in the future. Neoantigens are novel antigens generated by tumor-specific mutations that the immune system has never been recognized. These neoantigens are more advantageous than human antigens because they are unique to an individual tumor, they’re resistant to immune selection, and there’s a lower risk of self-tolerance and autoimmunity. Treatment with a neoantigen can help our immune system recognize and destroy the cancerous cells. Because each cancer is different, specific neoantigens must be made for each patient. The real question is: How are these personalized neoantigens manufactured?
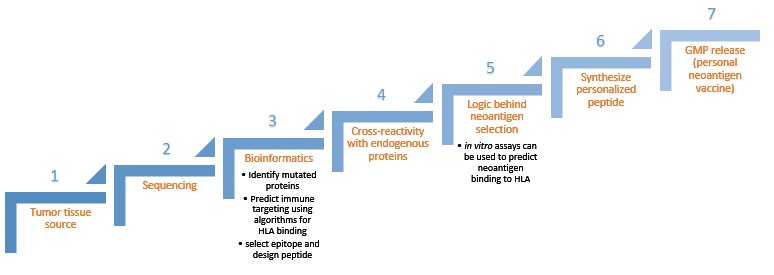
Syed Husain from FDA/CBER, Joel Greshock from Neon Therapeutics and Richard Bourgon from Genentech discussed ways to design and manufacture a personalized neoantigen product. The idea is that a tumor-specific mutated protein will be chopped in pieces and presented to the immune system in a way so that immune cells in your body can recognize the antigen and make antibodies against it. Identifying a high quality personal neoantigen becomes key for having a therapeutic effect, but empirical identification is not feasible. Therefore, the manufacturing steps (Figure 2) become important because individual peptides must be constructed so that they match the patient’s cancer mutations.
Comparability studies – how does manufacturing affect quality?
Comparability was the main area of focus during the third day of the conference. Comparability studies for proteins and biologics are essential in demonstrating that a manufacturing process change will not have an adverse impact on a biologic or biopharmaceutical product’s quality, safety or efficacy . Due to the limited starting material, narrow structure-function understanding, and restricted analytical toolbox available for cell and gene therapy products, comparability studies can be challenging. However, a risk-based approach and maintaining close communication with health authorities throughout product development can help overcome comparability challenges.
Cell and gene products are two great examples of potential therapies to treat and possibly cure many puzzling disorders. Open dialogues between industry, academic, and regulatory professionals around the world via conferences like this one support the advancement of the manufacturing process of these products, which assists the medical practice community and patients suffering from debilitating diseases.
This conference also offered a wonderful opportunity for young professionals such as postdocs and graduate students to network, fostering valuable connections. If you are a graduate student or postdoc at Johns Hopkins interested in a professional career outside of academia, the Johns Hopkins Professional Development and Career Office has partnered with CASSS to match graduate students with mentors to meet monthly for six months with the goal of mentoring students on transitioning to careers in industry or government. The CASSS-Johns Hopkins University Mentor Match Program helps students establish a relationship to discuss career interests, resumes and skillsets, elevator pitches and current trends in fields of interest to become exceptional candidates for industry jobs. The CASSS-JHU Mentor Match Program enrolled a new cohort of students in the fall, and the program will likely continue in the spring.
Graduate students or postdocs at any university can meet their mentors in person by qualifying for CASSS’ student travel grant, which waives the conference registration fees. Each conference has specific application requirements (e.g., write a synopsis of the conference or present a poster) — please read more by visiting the CASSS website.
Related content
- Advancing the Manufacturing of Human Cell and Gene Therapies
- Collaborations with Industry Strengthen Academic Research
- Insights from the Rock Health Enterprise Insights Series
Want to read more from the Johns Hopkins School of Medicine? Subscribe to the Biomedical Odyssey blog and receive new posts directly in your inbox.