
Last November, I was able to attend the National Science Policy Network Symposium in Madison, Wisconsin. The goal of this two-day symposium was to understand how many scientific issues (such as climate change and genome-based medicine) disproportionately affect minority and vulnerable populations. One of the workshops I attended discussed how “big genomic data” is handled, and how many of the algorithms and statistical methods used nowadays can perpetuate already established prejudices. This was shocking to me, as I had previously thought of algorithms as inherently unbiased. How can math perpetuate inequality?
Our genome is extremely complex. Many of our traits, as well as risks for disease, are determined by a combination of many genes as well as by interactions with the environment. Therefore, any associated risk to develop a certain disease is polygenic.1 Each of the many genes contributing to a specific disease has the ability to increase or decrease the chances of developing it. Scientists can use the association of different gene variations with certain diseases to predict the likelihood that an individual will develop a disease via genomewide association studies (GWAS).
How does GWAS work?
Our genome is built of 3 billion nucleotide base pairs: That is a lot! GWAS looks at single nucleotide polymorphisms, also known as SNPs, which are parts of the genome in which one base is switched to another.2 These SNPs are random and aren’t per se harmful. But they do provide the basis for us to ask questions and try to find associations. Are there specific SNPs that happen in people who suffer from diabetes? What about breast cancer? GWAS, therefore, provides correlations between SNPs and a specific trait or disease. However, it is important to remember that these are just statistical associations and aren’t by any means causative. That is to say, just because an individual has a particular SNP that is associated with a disease doesn’t necessarily mean they will develop that disease.2
The effect each of the SNPs has on a predictive risk is extremely small, so scientists combine all of the SNPs found in someone’s genome to produce a “polygenic score” for a certain disease or trait, which has a lot of predictive power. Although this technology is extremely useful and has the power to do a lot of good, GWAS can be misused in ways that perpetuate health disparities.
GWAS Limitations
As you might imagine, it takes huge datasets for GWAS to correlate specific base changes to traits and diseases. The more studies performed, the more correlations will be found. However, GWAS studies are limited in that the majority of them have been performed in populations of European background.3 This is important, since different populations might acquire random genomic variations, and predictions from these studies cannot be used to predict disease risk in other populations. So if you’re not from a European background, you do not benefit from these new predictive technologies.3 This reinforces our preexisting problem that people of color are often not included in medical research studies, which, in turn, could aggravate medical disparities in America. It actually already has. A recent study by Manrai et al.4 showed that patients from African or unspecified ancestry were misdiagnosed with hypertrophic cardiomyopathy based on their genetic testing results. This occurred because the mutations used to identify a patient to be at risk for the disease were far more common among African Americans than white Americans. For African Americans, that set of mutations does not have strong predictive power for developing hypertrophic cardiomyopathy. For this reason, it is essential that GWAS include people from ancestry groups that are understudied, such as African ancestry.
As shown in the graph below,5 people with European ancestry are overrepresented in GWAS studies. Most of the GWAS data that scientists have gathered comes from individuals with European background, even though these individuals account for a way smaller portion of the world population. If we want to make progress in precision medicine and we want these studies to be an effective and equitable tool, GWAS needs to be performed in more diverse populations. So why aren’t we doing this?
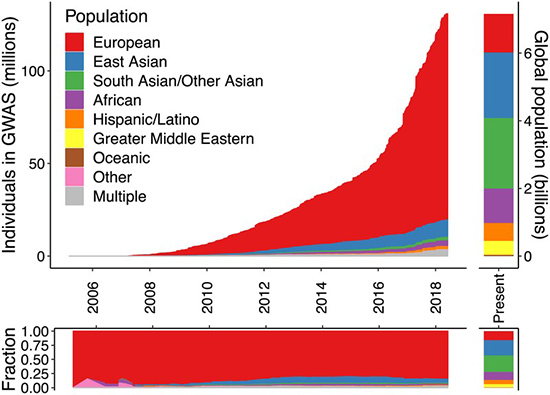
Figure 1. Ancestry of individuals who participated in GWAS studies compared with world’s population.
To make a GWAS study accurate in predicting biological outcomes, the pool of participants needs to be from a common ancestry. This is done to prevent possible wrong associations due to population stratification. In other words, if GWAS studies included people from mixed ancestries, they might find associations between gene variants and diseases that could be explained simply by a specific population having a higher frequency of that gene variant. One clear example of this was explained by Campbell et al,6 which found an association between an SNP in the LCT gene (responsible for the correct digestion of lactose) and height. This would suggest that taller people have a higher risk of developing lactose intolerance. We know, of course, that this isn’t true. When researchers reevaluated the individuals in conjunction with their ancestry, they found that this association was a consequence of stratification and that no real association was present.
This explains why GWAS study cohorts are pretty homogenous, but doesn’t explain why we haven’t done more GWAS studies in populations coming from non-European ancestries in order to have GWAS data that provides reliable results for every person. One reason might be the lack of diversity in the scientific field.7 Researchers and investigators might have personal connections and personal interests in pursuing a specific study, which could result in the study cohort mirroring the homogeneity of the scientific field itself. Therefore, the underrepresentation of low-income and minority groups in the scientific field can impact the type of communities served by the research. Another reason is the “preferred cohort” effect.7 This refers to the idea that, because historically much of the genomic research has been done in cohorts of European ancestry, there are pressures for researchers to keep their research focused on this population. Reviewers and editors who make funding and publication decisions expect researchers to build off these historic studies, and so may favor studies that use a similar population. Lastly, due to population structure and migration patterns, it is usually easier to accurately associate genetic variants with disease risk in these European populations. All of these factors have led to the development of a body of GWAS data that disproportionately serves individuals of European descent.
Remaining Questions
GWAS results allow us to make predictions based on just a percentage of our genome, but these studies could worsen existing health disparities. A lot of questions remain open regarding the impact that GWAS and risk scores will have for everyone. One of these is whether insurance companies will require access to a predictive score that could then be used as a “preexisting condition” and grounds to deny insurance coverage. In addition, many private companies such as 23andMe offer a range of personal genotyping options to learn your associated risk to develop common diseases based on your ancestry using data generated by GWAS studies. The accuracy and efficacy of these services for specific populations has yet to be studied. We do not know what the ramifications of improper GWAS data regulation will be, but as with all powerful technology, care must be taken to ensure the technology can serve all populations equitably.
References
- “Polygenic Risk Scores.” gov.
- “Genome-Wide Association Studies Fact Sheet.” gov
- Genetics for all. Nat Genet51, 579 (2019).
- Manrai, Arjun K., et al. "Genetic misdiagnoses and the potential for health disparities." New England Journal of Medicine7 (2016): 655-665.
- Martin, Alicia R., et al. "Human demographic history impacts genetic risk prediction across diverse populations." The American Journal of Human Genetics4 (2017): 635-649.
- Campbell, Catarina D., et al. "Demonstrating stratification in a European American population." Nature genetics8 (2005): 868-872.
- Bentley, Amy R., Shawneequa Callier, and Charles N. Rotimi. "Diversity and inclusion in genomic research: why the uneven progress?." Journal of community genetics4 (2017): 255-266.